Machine Learning for Natural Sciences
Decision trees, ensemble methods
Decision tree construction algorithm
- For each input dimension (k) and each cut along that dimension (n), select the cut with minimum impurity
- Recursively repeat that for both subtrees until stopping criterion is reached
Measures for purity
Minimum Entropy
Minimum Classification Error
Gini-Index
Runtime analysis of decision tree construction ❓
For each split, we need to test k·n possible splits. For sorting the splits to get the best possible one, we have a complexity of n·log(n). Therefore, the runtime to train the root node scales with k·n·log(n) The average depth of the tree is log(n), because each split halves the size of the samples, leading to pure nodes with single samples after log(n) decisions. Therefore, the overall scaling to train all log(n) levels is the product of both, i.e. k·n·log(n)*log(n)
Regularization methods for decision trees
- Early stopping when the impurity reduction is lower than a threshold
- Early stopping based on cross validation
- Early stopping for minimum number of nodes per samples
- If composite function does not go down anymore
Problems with decision tree regularization
- Hyper-parameters
- Early stopping has only current information
- Better: Post-Pruning: First construct full tree and prune tree afterwards
What kind of data causes problems for decision trees?
Correlated features
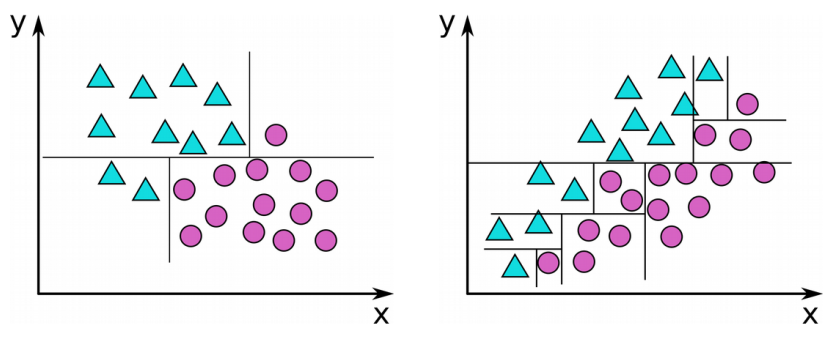
What can be done about that?
Use polythetic decision trees: Look at splits that depend on multiple variables
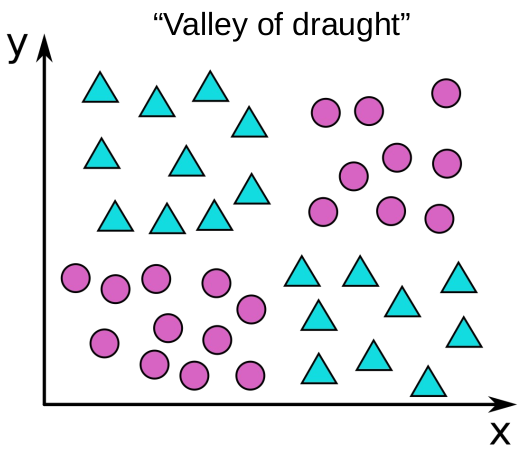
Which problem can arise when using information gain as a stopping criterion for decision tree training? Describe a dataset for which the problem occurs.
“Valley of draught”: In one split the impurity can’t decrease, even though the data is separable along the features.
What ensemble methods are there?
- Bagging (Bootstrap aggregation): Train independent models on random subsets of the data in parallel
- Boosting: Train models sequentially. Later models focus on the missclassifications of the model before.
Definition Bias
Definition Variance
Bias-Variance Tradeoff
Random split selection
- Do not offer all features for split at a node
- A combination of weak nodes can result in strong features
Derivation variance reduction for bagging (random forests)
It is well known that
So we can show
Advantages and disadvantages of random forests
- Can reach high accuracy with a low number of parameters (v.s. neural networks)
- Works for categorical and continuous data
- Fast training and prediction
- Scaling invariant
- Interpretability, in the sense that it can assign importance to features
- Intrinsic estimation of generalization error
- Need handcrafted features
Intrinsic estimation of generalization error in random forests
- In-bag samples are used for training
- Out-of-bag samples are used for error estimation
How to measure feature importance
Gini importance
- Sum of the reduction of impurities of nodes of given feature
- Sum this over all decision trees and weight by the number of samples in the node
- But: Categorical variables are preferred
Permutation importance
- Permute values of out-of-bag samples at a given feature and measure increased error
- Problematic for correlated data
Conditional permutation importance
- Improvement of permutation importance
- Only permute, if between samples with similar values for other features
What can decision trees be used for?
Classification of cosmic rays
- We want to detect interesting cosmic events live supernovas and formation of black holes
- These cause gamma-ray explosions, that are the brightest known events in the universe
- Such gamma rays can be detected indirectly
- Gamma-rays travel at the speed of light through the universe
- When entering the atmosphere, they slow down
- Therefore, energy is released in form of Cherenkov radiation, which are photons
- From the image data, features can be extracted
- With the features, a random forest can be trained to distinguish between gamma-ray explosions and other events
Distinguish benign and malevolent breast cancer tumors, i.e. which tumors are lethal
- Use eight features (tumor size, age, …)
- Problem: No binary labels, instead use so-called survival tree, which uses the lower limit of survival time
- Estimating feature importance gave new insights
Prediction of protein interaction
- It is interesting to know which surface proteins bind together
- Train a random forest to predict if two proteins bind well together
- Feature importance is used to reduce the feature space and train better models, as well as giving insights for other research
Catalysis
- Catalysts enable/accelerate chemical reactions
- Predict the reaction barrier of different catalysts
- Use fingerprint vectors of molecules as input
- Use feature importance to find important properties of the molecule
- Test the hypotheses automatically
How can you interpret the predictions of a random forest? Explain two approaches.
- Feature importance
- Looking at small sample of random forests and their decision for a single instance
Mathematical foundations
Are not explicitly asked in the exam, but still important to know for their applications
norm definition
Properties of norms
Max norm
Frobenius matrix norm
Scalar product expressed as norms
Bayes’law
Goal of PCA
Map m inputs of dimension n: with minimal information loss
Encode:
Reconstruction:
Derivation of the compression and reconstruction function of PCA


Kullback-Leibler (KL) divergence
Application of PCA for natural science
Project to lower dimensional space in order to visualize and see clusters and outliers. E.g. molecules, cell data or particle accelerator data
Machine learning basics
Learning algorithms - definition
“A computer program is said to learn from experience E with respect to some class of tasks T and performance measure P, if its performance at tasks in T, as measured by P, improves with experience E.“ Mitchell 1997
Important Tasks T

Formulation of unsupervised problem as supervised problem
Formulation of supervised problem as unsupervised problem
MSE
Linear regression solution derivation
Linear regression as a maximum likelihood problem
Bayesian learning
Compute Posterior:
Compute Predictive Distribution
Maximum-a-posteriori estimation

SVM classification and optimization

SVMs for non-linear data
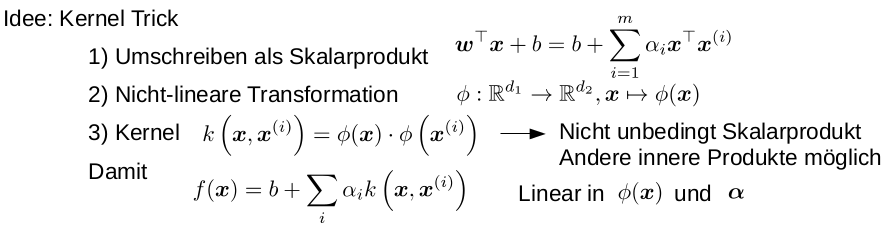
TPR, FPR, confusion matrix
ROC curve
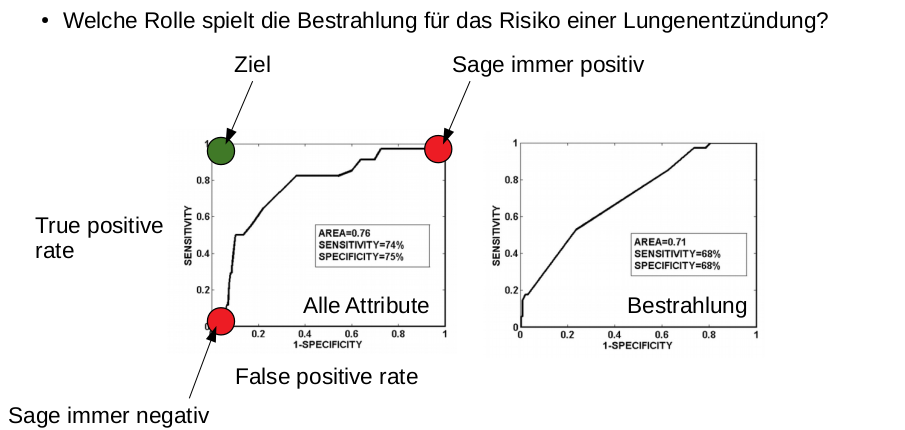
k-means algorithm
- Chose k
- Start with k (random) vectors (cluster centers)
- Iterate
- Assign each data point to it the nearest cluster center
- Move the cluster centers to the mean value of the respective groups/clusters
Idea of manifold learning
Only consider data on a manifold, e.g. by projection
Neural networks, deep learning
logistic regression
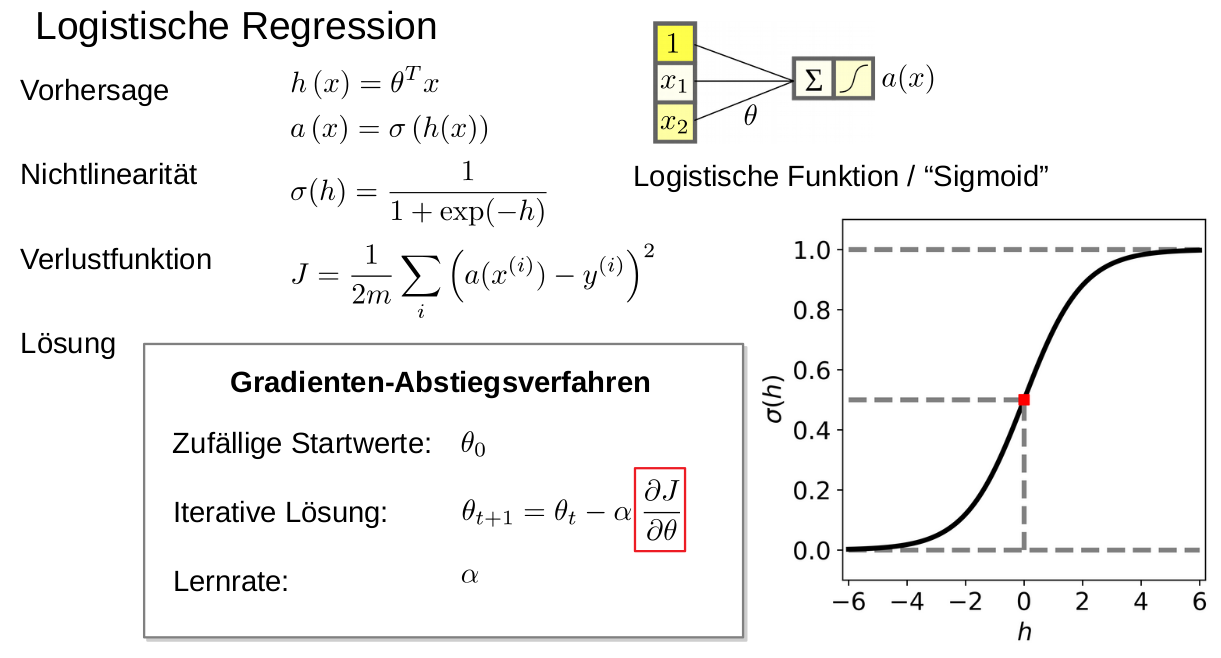
Logistic regression backpropagation

Regularization of neural networks
For neural networks, often only regularize the weights and not the bias, there are much more weights than biases
Downsides of regularization by data augmentation
- Invariance must be known
- But in this case very useful
- Not applicable to all tasks
- Invariance must be known
Multitask Learning
- Learn common first layer for different applications
- Some may even be unsupervised
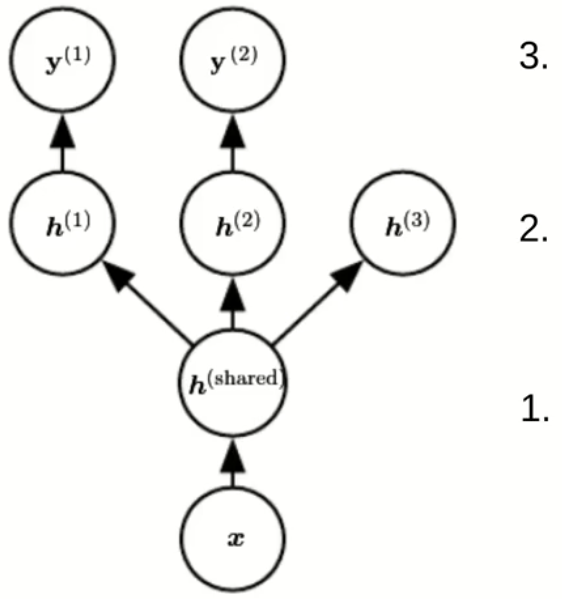
- Similar to fine-tuning
- Learn common first layer for different applications
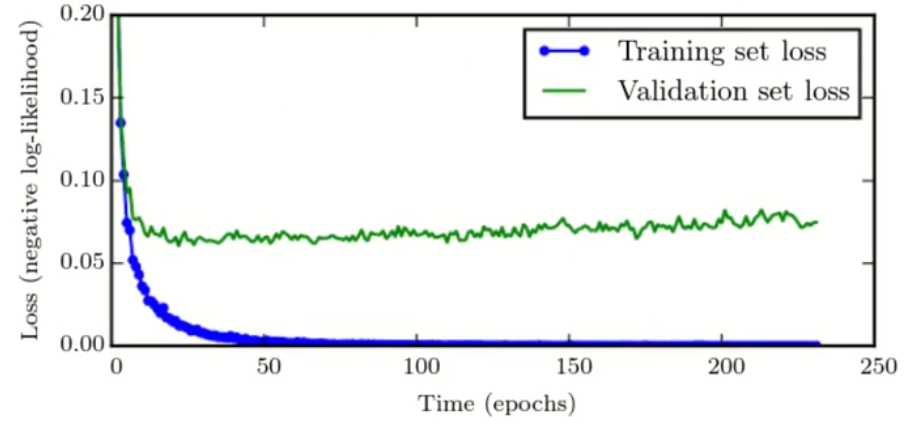
Early stopping
Stop if validation error does not improve for some time
Similar effect than L2 regularization
Parameter sharing
- Enforce equality of parameter at different positions
Give an example where parameter sharing is used
Most famous example: Convolutional neural networks
Sparse regularization
- Penalize non-zero entries of representations (instead of weights as seen in L1 regularization)

How should the loss curves of a model change after adequate regularization?
The training error to be a little higher and the validation loss is expected to decrease further and rise later.
Draw the (expected) ROC curve of a classifier that outputs uniformly random predictions in a two class setting. Do not forget axis labels
Straight line trough (0,0) and (1,1). Y-axis FPR, X-axis TPR
Let . Show that maximizing the likelihood is equivalent to minimizing the MSE.
Just calculate the log likelihood of the density function and then it is easy to see that the maximum is equal to the minimal MSE.
Why is feature selection particularly relevant for k-NN?
To avoid the curse of dimensionality and eliminate noise dimensions that hurt the model, because it does not weight dimension.
Atomic-resolution simulations with neural networks
Give a few examples of nanoscale phenomena that can be simulated.
Systems with nanoscale functional units, for example:
- Biology
- Biological processes in cells
- Proteins and DNA
- Drugs and their function
- Chemistry
- Chemical and catalytic reactions
- Technology
- Fuel cells and batteries (e.g. Li-Ion)
- Solar cells and displays
- Electronics and semiconductors (e.g. for electronic circuits)
- Biology
What kind of forces and interactions occur in a multi-particle system?
- kinetic energy of electrons
- kinetic energy of nuclei
- Electron, nucleus interaction
- nucleus, nucleus interaction
- electron, electron interaction
Name three classical methods for atomic resolution simulation
- Simulate only atoms with classical mechanics, ignoring quantum physical effects
- Empirical force fields
- Density Functional Theory (DFT)
If you have a network predicting the energy of a system, how can you make a next step prediction of the particle positions?
Why use machine learning for atomic simulations?
DFT calculation are computationally expensive. to more precise variants with to .
Machine learning can give similar accuracy with less computational complexity.
Machine learning learns the potentials. So we have a mapping from the molecule coordinates to the energy.
What is the problem with representing a molecule as a vector of inverse distances between its atoms?
This is molecule specific → Cannot be transferred between molecules
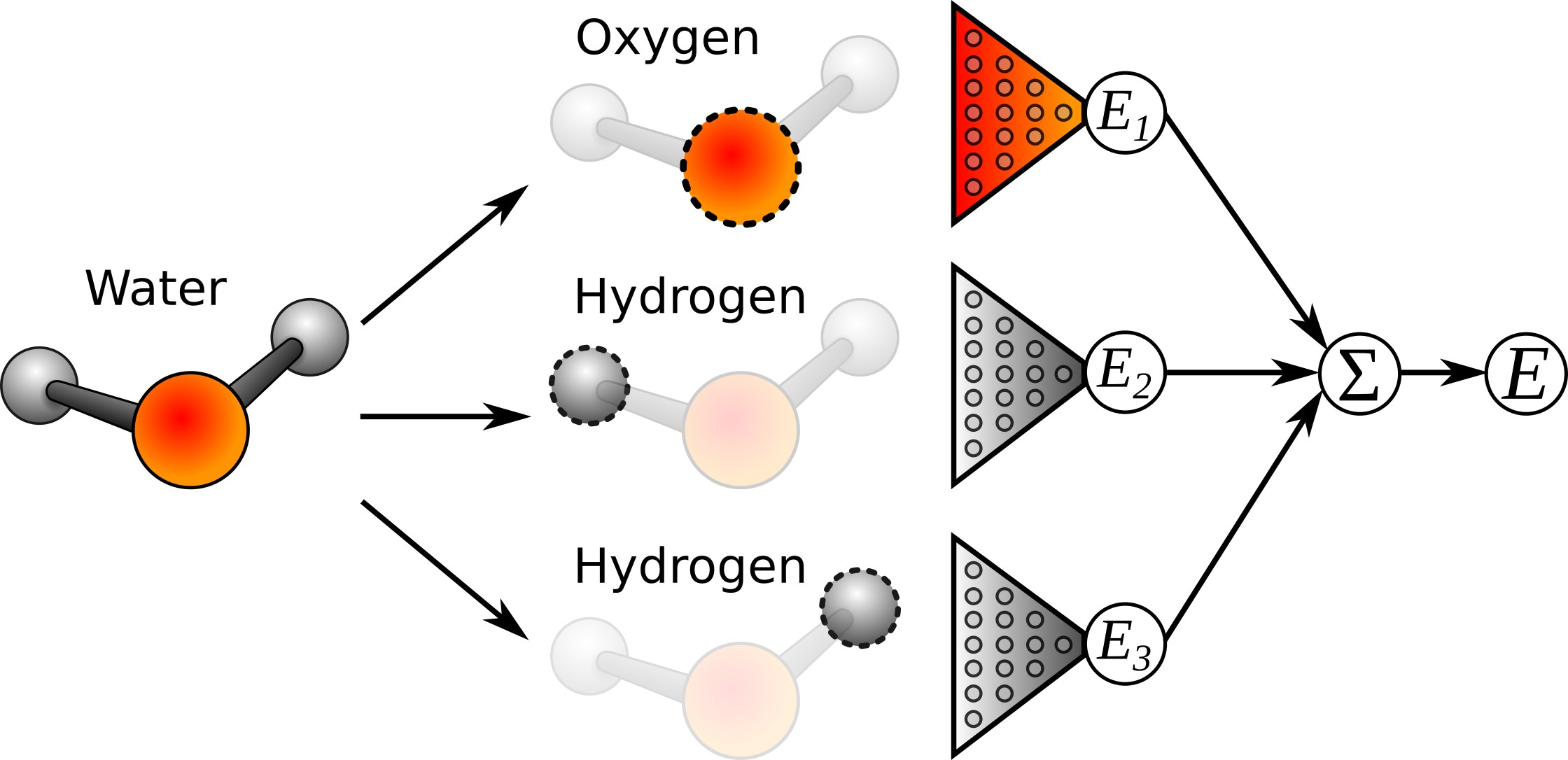
Draw an architecture for learning the energy of a molecule
- Subdivision of the problem into local subproblems, typically per atom.
- Both hydrogen atoms have the same neural network, but the input vectors are different because their environment differs (they are not perfectly symmetrical)
How to create off-equilibrium molecule datasets for atomic simulation? Explain two approaches and compare them.
Algorithmic creation of geometries
Vibration-based
- Calculate equilibrium geometries
- Calculate Hessian matrix and its eigenvectors (eigenmodes/oscillations)
- Distort/move the atoms using random linear combinations of these eigenmodes
Trajectory-based
Calculate MD trajectories of single molecules at given simulation temperatures (ideally higher than target temperature) Take a fraction of the MD steps for training, due to redundancies in subsequent steps
Comparison
- Vibration-based
- Problem: Hessian matrix is only a harmonic approximation
- Dihedral rotations are not sampled well → Conformer search
- Trajectory-based
- Problem: Highly correlated data
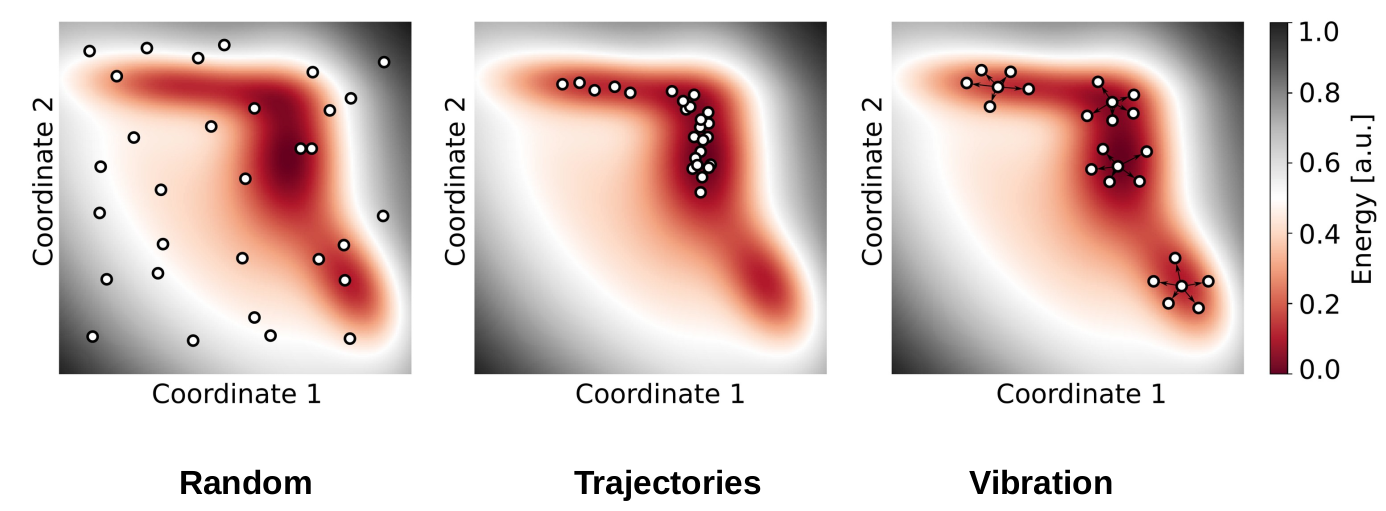
- Vibration-based
CNNs and medical image data
Definition 1D convolution
Definition 2D convolution
Definition cross-correlation
CNNs: Invariances
- CNNs are not invariant to rotations
- Slightly invariant to scaling
- Slightly invariant to translations
How to deal with the problem of bounding boxes in the background when using a one-step method

How does the presented model for breast cancer detection work?
- The first model detects suspicious regions. And classifies if there is cancer in it or not
- The second model makes independent predictions for the left breast and the right breast. For each breast, there are multiple pictures. These are concatenated as channels. With ResNet on the full images. Only the results are combined
- The third model makes a prediction based on all images, also using ResNet. Concatinating images after they went trough resnet.
Draw a ResNet layer
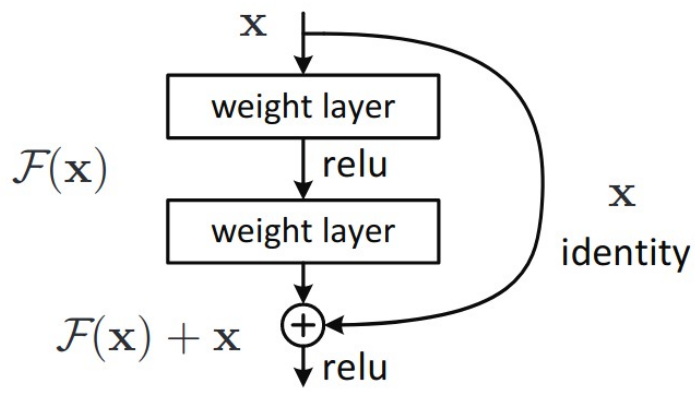
Idea residual connections
- There are direct connections from the initial layers to the output layer and thus the loss function.
- This prevents the problem of vanishing gradients and enables the (more efficient) training of deeper architectures.
- Intuitively, this learns corrections of the input, giving the fully connected layer a useful representation that includes both higher level features and lower level features.
CNNs for quantum chemistry

What architecture can be used for segmentation of cells
U-Net

Explain this architecture
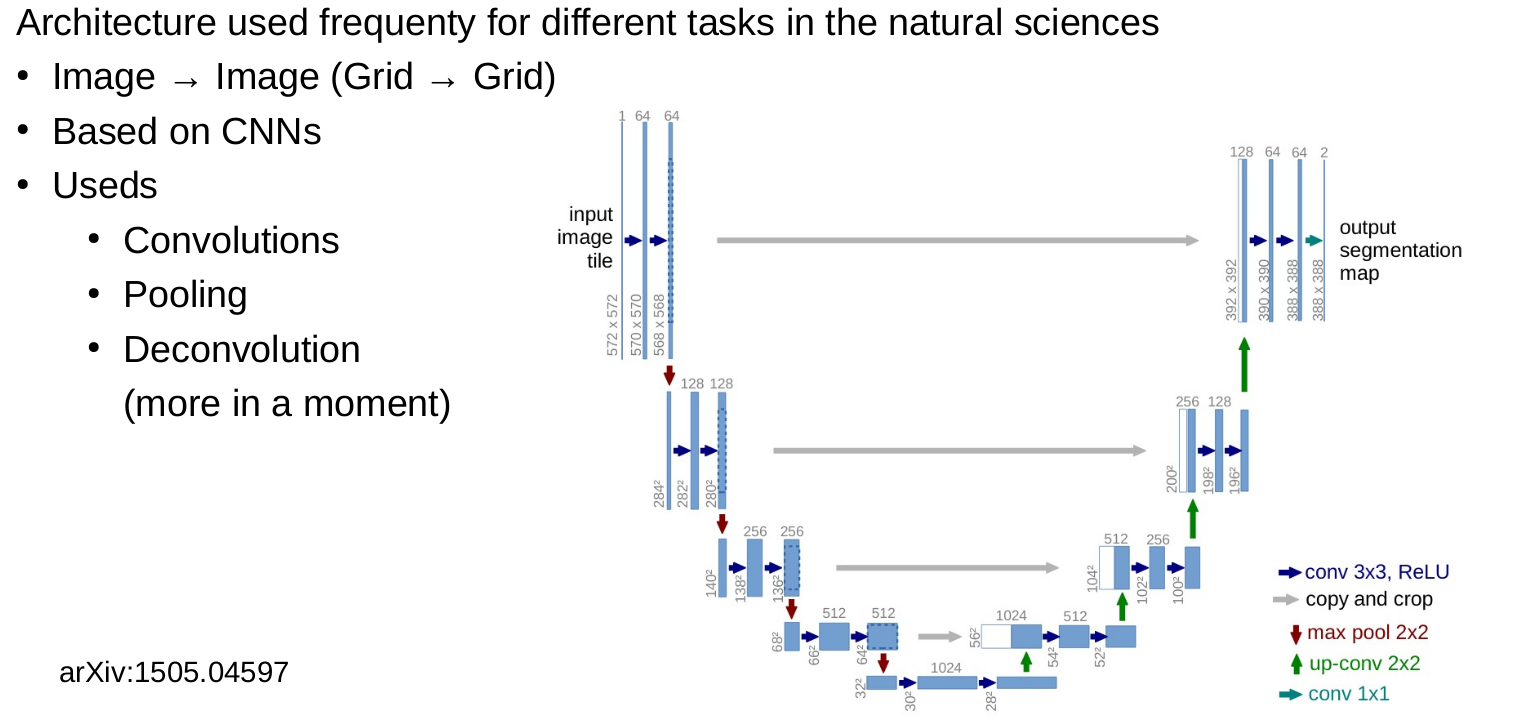
Draw an example deconvolution
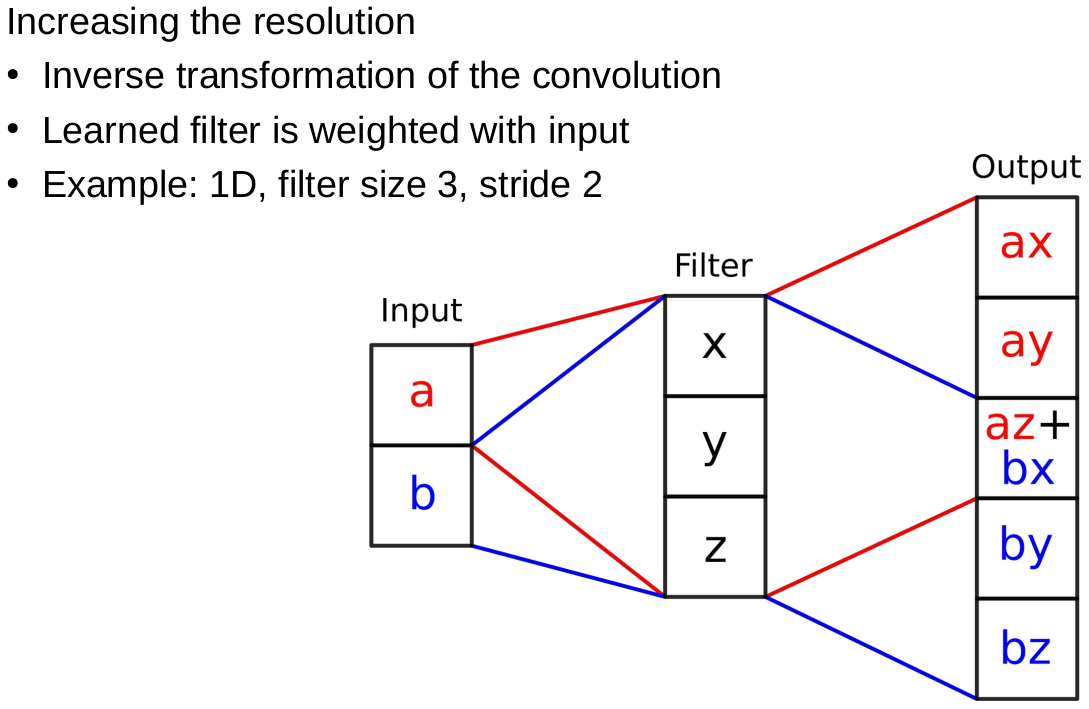
What is the difference between ResNet v1 and ResNet v2?
ResNet v1 applies the activation function to the skip connection as well (after the addition)
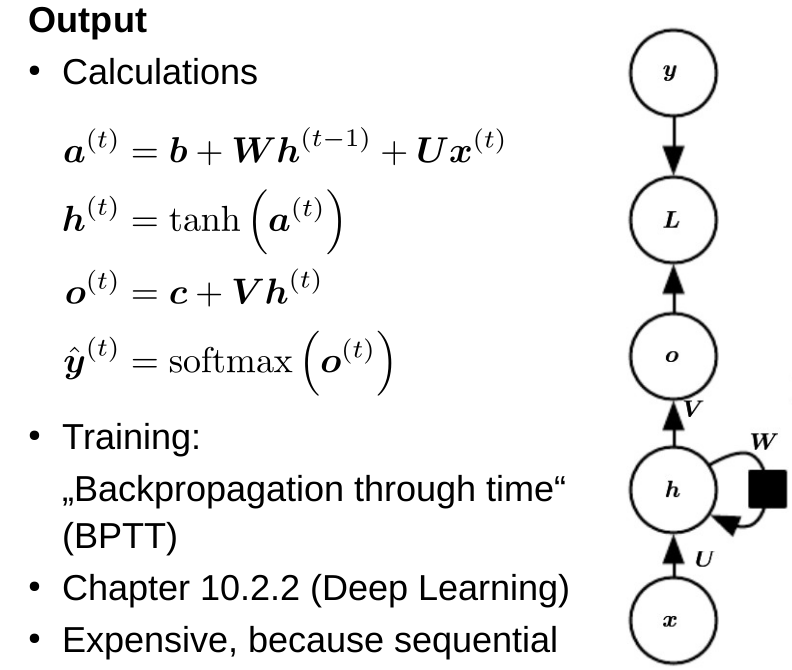
RNNs and reaction prediction
Equations for RNN training
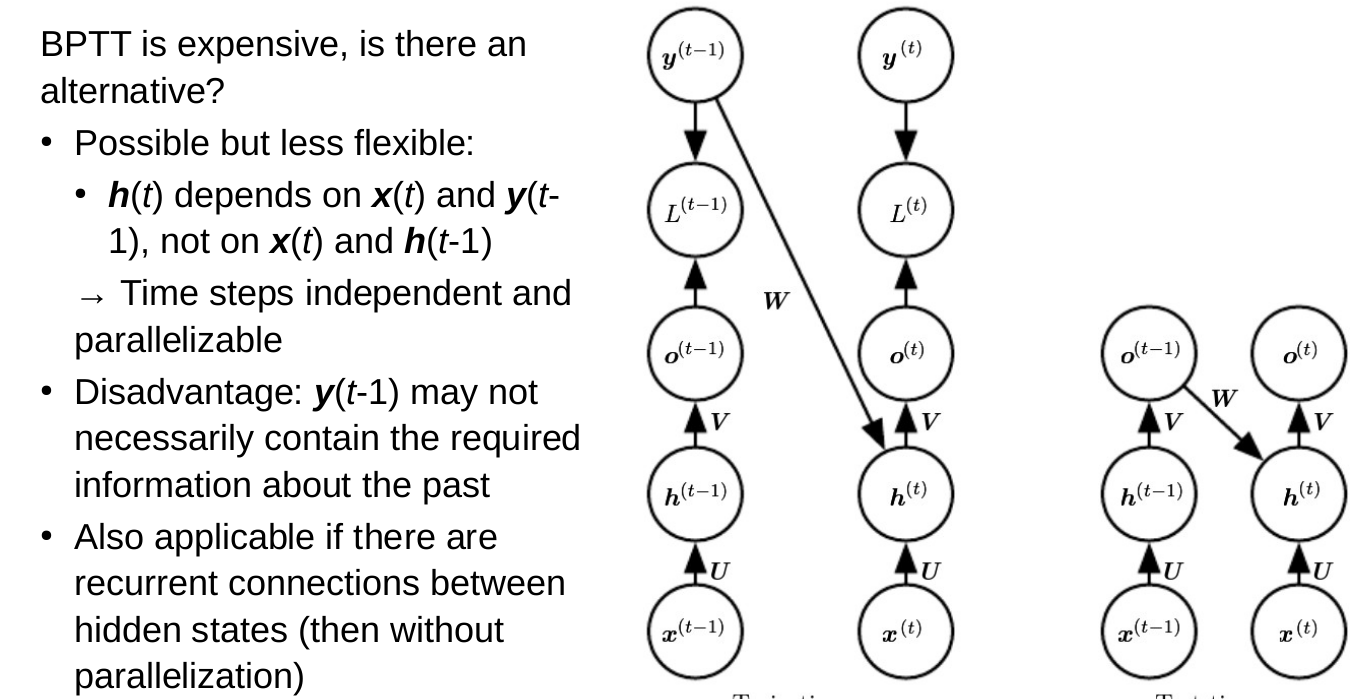
BPTT is expensive, is there an alternative?
Teacher forcing
Draw a sequence to sequence model
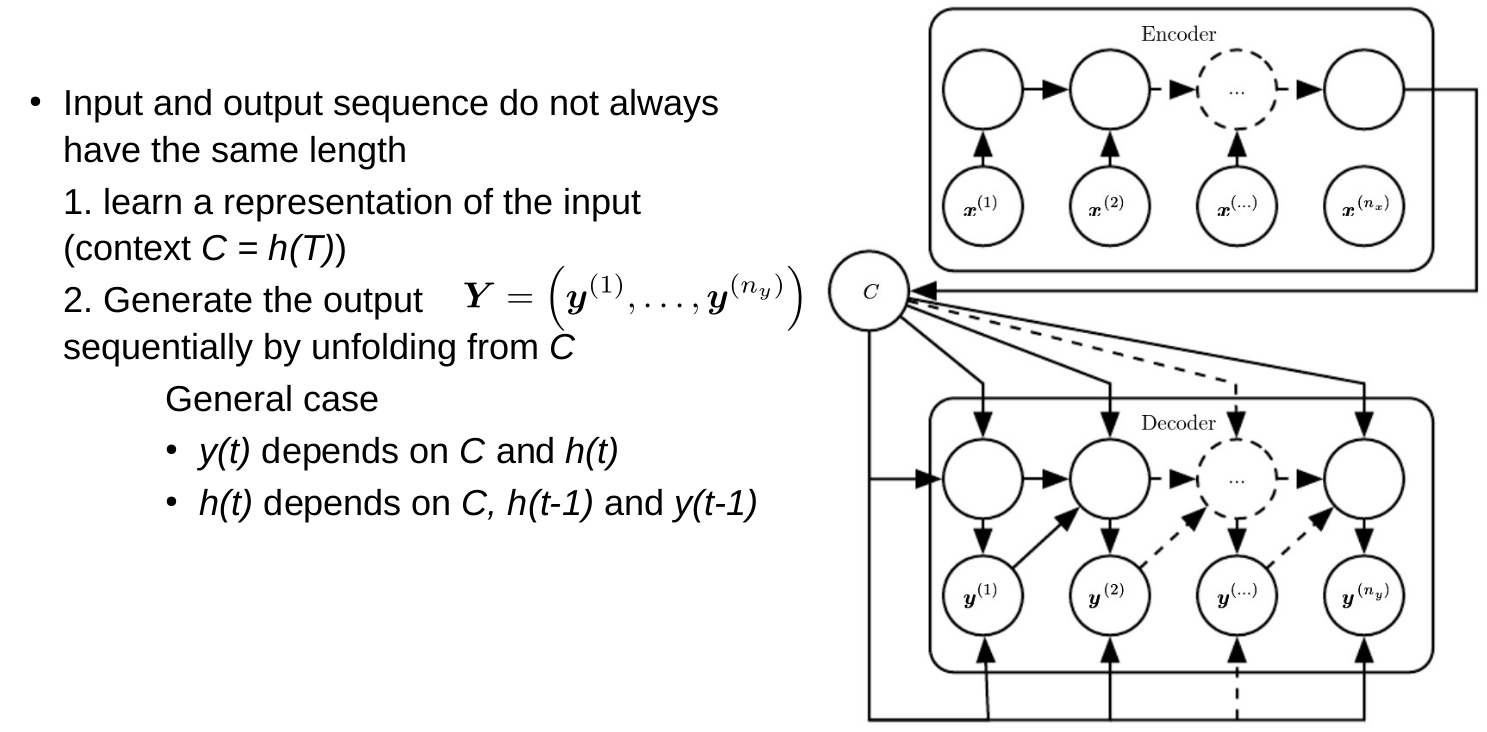
Similarity between sequence to sequence models and autoencoders

Options for deep RNNs
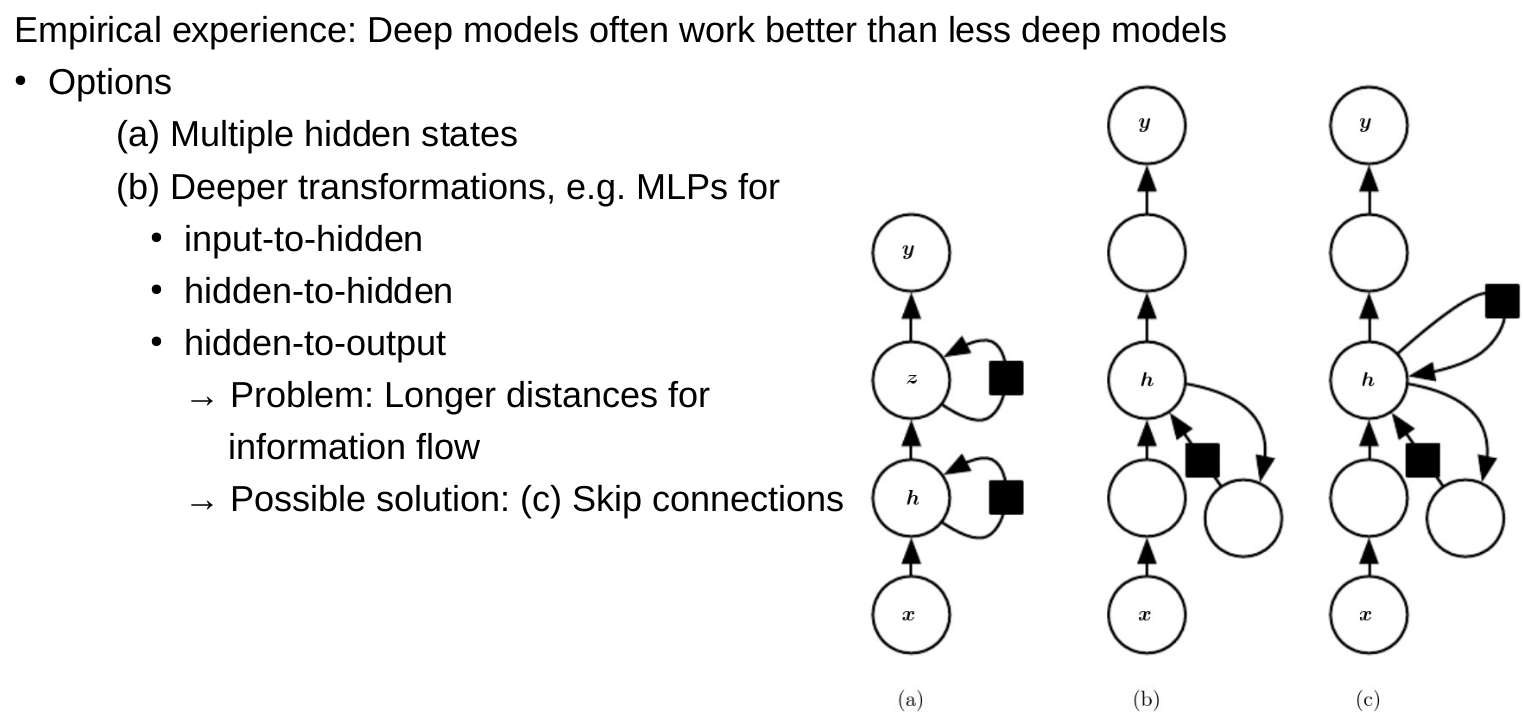
Problem of sequence to sequence
- Everything must be stored in a hidden state (context)
- You can't look back
- Example: Read a book, then get asked a question
What are the components of a LSTM?
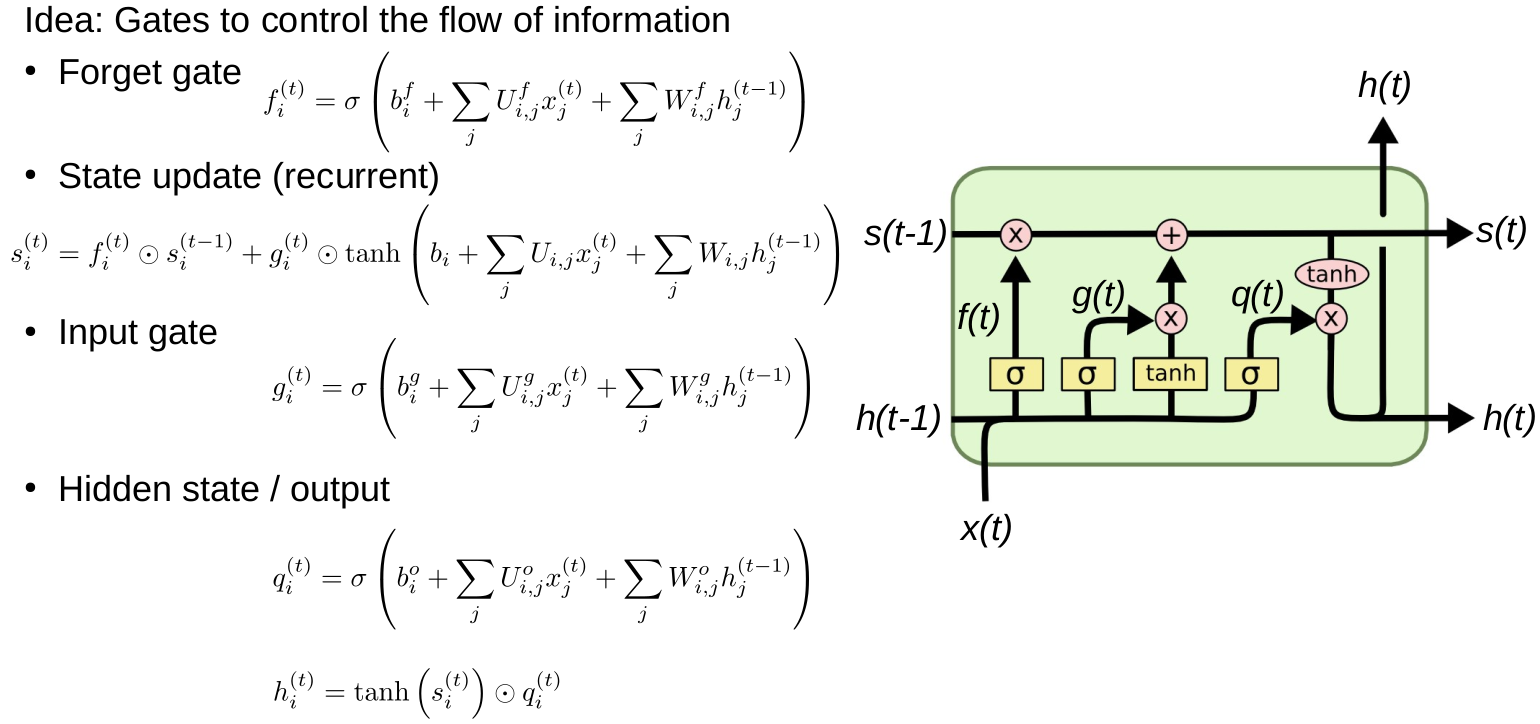
What is the idea of attention?

How does an alignment for automatic translation matrix look like?

RNN applications
Analyzing electrocardiogram data
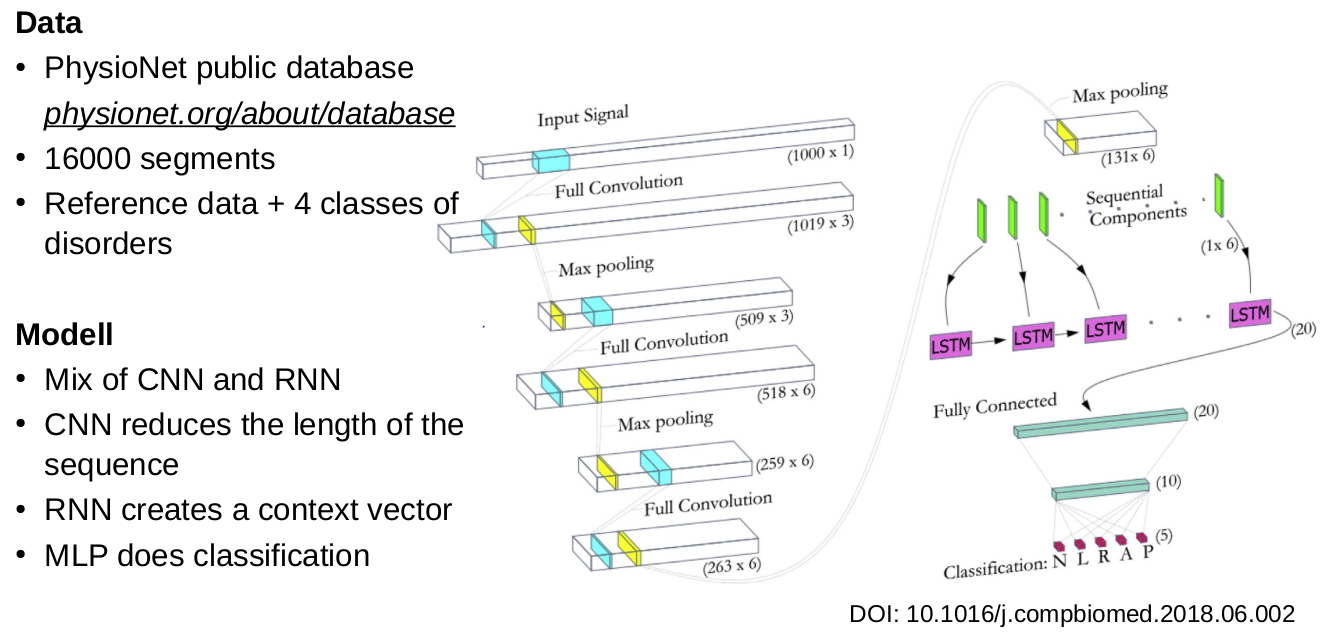
Jet classification: Finding Proton-antiproton collisions
- Input: time series of measurement
- Output: proton-antiproton collision or not
- Training done via simulations
- Good results with LSTM
Predicting the results of chemical reactions
- Input: starting molecules as SMILES code
- Output: Products of the reaction as SMILES code
- Model: Transformer
Graph neural networks and molecular representations
Draw the molecule O=C(O)C(N)CC1=CC=CC=C1
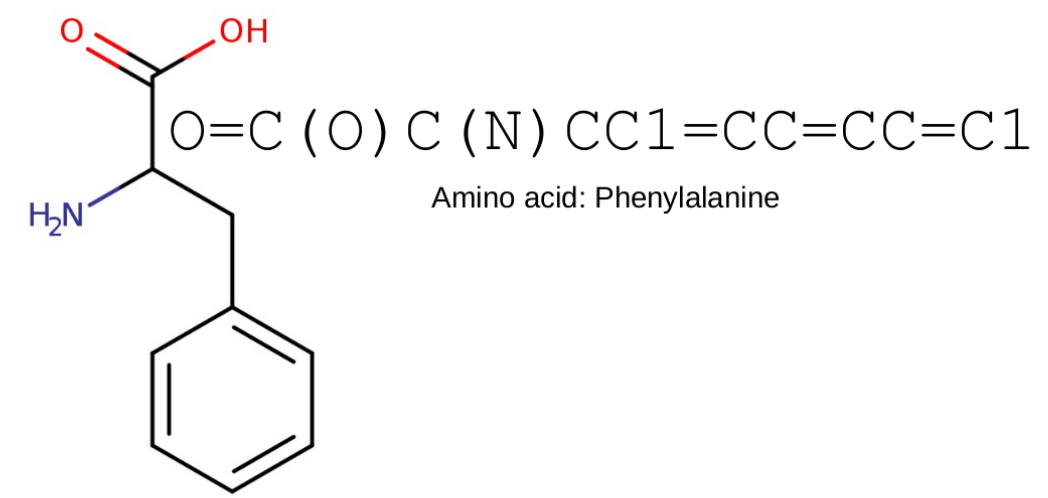
Advantages and disadvantages of smiles code
- Advantages
- Human-readable
- Can represent any molecules
- Relatively simple
- Disadvantages
- Not robust: Small change, big effect
- Not every string is valid
- Syntax is at least a context free grammar (because of parentheses)
- No “natural” representation of molecules
- Advantages
Basic fingerprinting of molecules
- Representation in the form of bit vectors
- Each bit describes the presence of a particular subgraph
- Each bit describes the presence of a particular subgraph
- Interpretable
- There are millions of possible subgraphs (with a given maximal size)
- They need to be identified and mapped to a bit vector of e.g. 1024 bit length
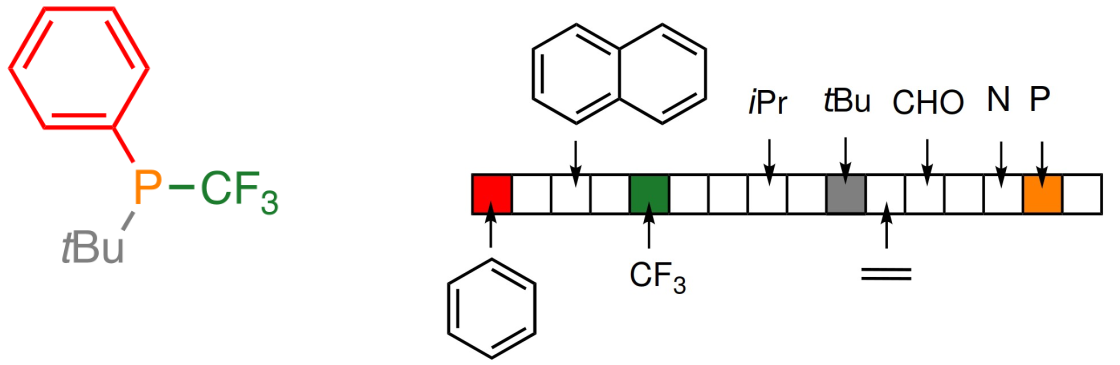
Extended-Connectivity Fingerprints
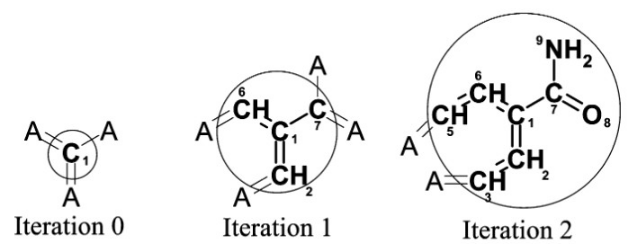
- Iterative generation of molecular representations that are as unique as possible
- Each atom receives an identifier
- In each iteration step the identifier is renewed, by aggregation with the neighbors
- The number of iteration steps describes the level of detail of the fingerprints („radius“)
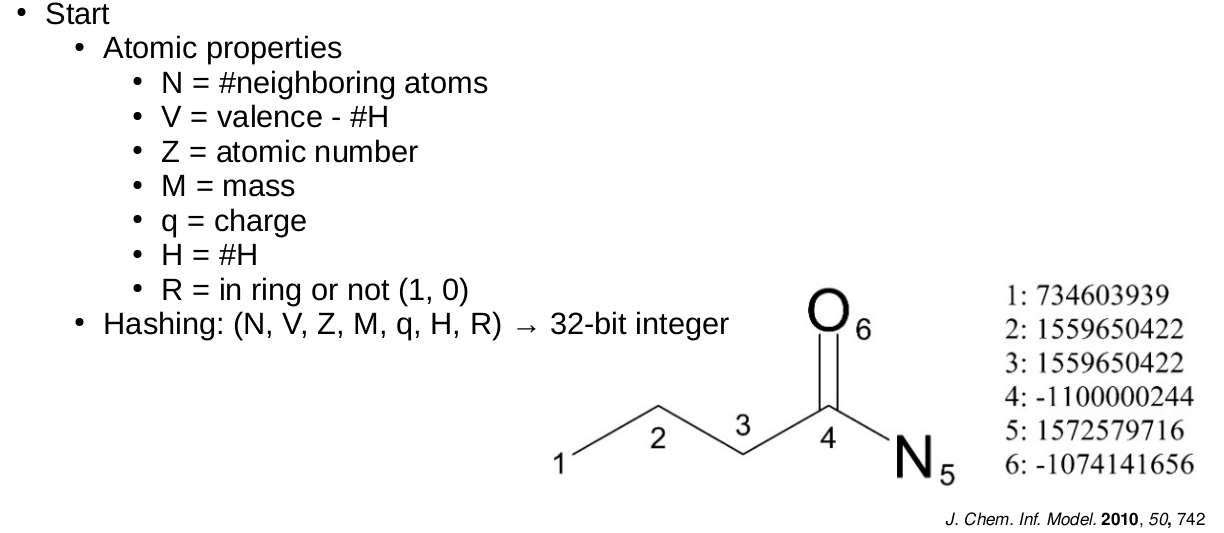
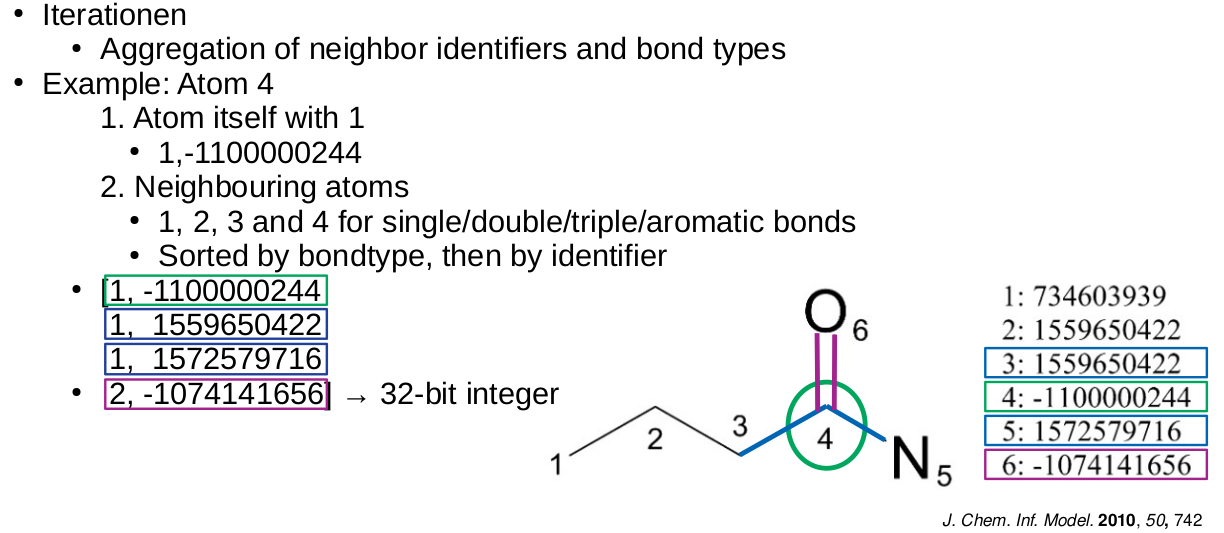
Hashing and aggregation inputs for Extended-Connectivity Fingerprints
How can graph neural networks be used for representing molecules?
Learn real valued vector similar to extended connectivity fingerprints (2015)

Message Passing Neural Network, 2017
How is the graph constructed, initially?
- Nodes are the atoms. Each atom has a state vector
- Edges are atom bonds. Each edge has a state vector
In each iteration, each node sends a message to each of its neighbors. How is a message from to calculated?
How is calculated?
How are the node states updated?
How is the final representation obtained?
The fingerprint vector of the molecule is obtained via the readout function
What can we do with such learned representation
Feed through a multi layer perceptron, to predict properties, such as toxicity or solubility
Autoencoders and generative models
What problems occur when using just the loss function for a vanilla autoencoder. How can this problem be fixed.

What is the learning objective of a contractive autoencoder? Write down the formula and explain the intuition behind it.
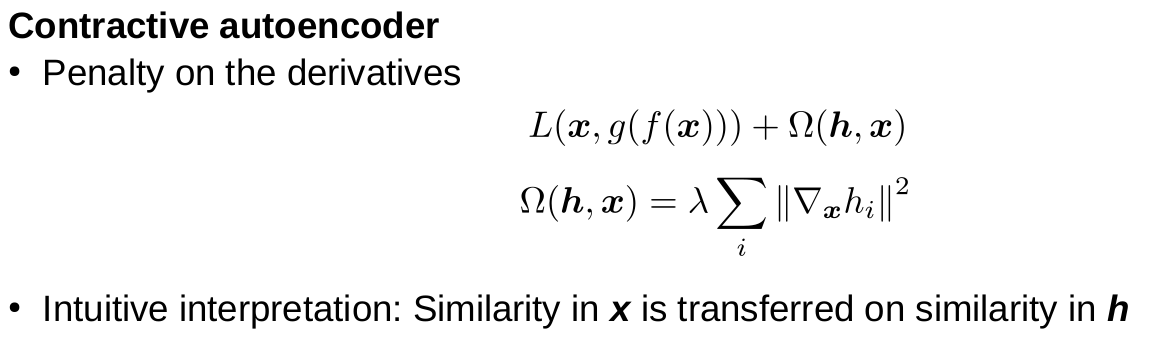
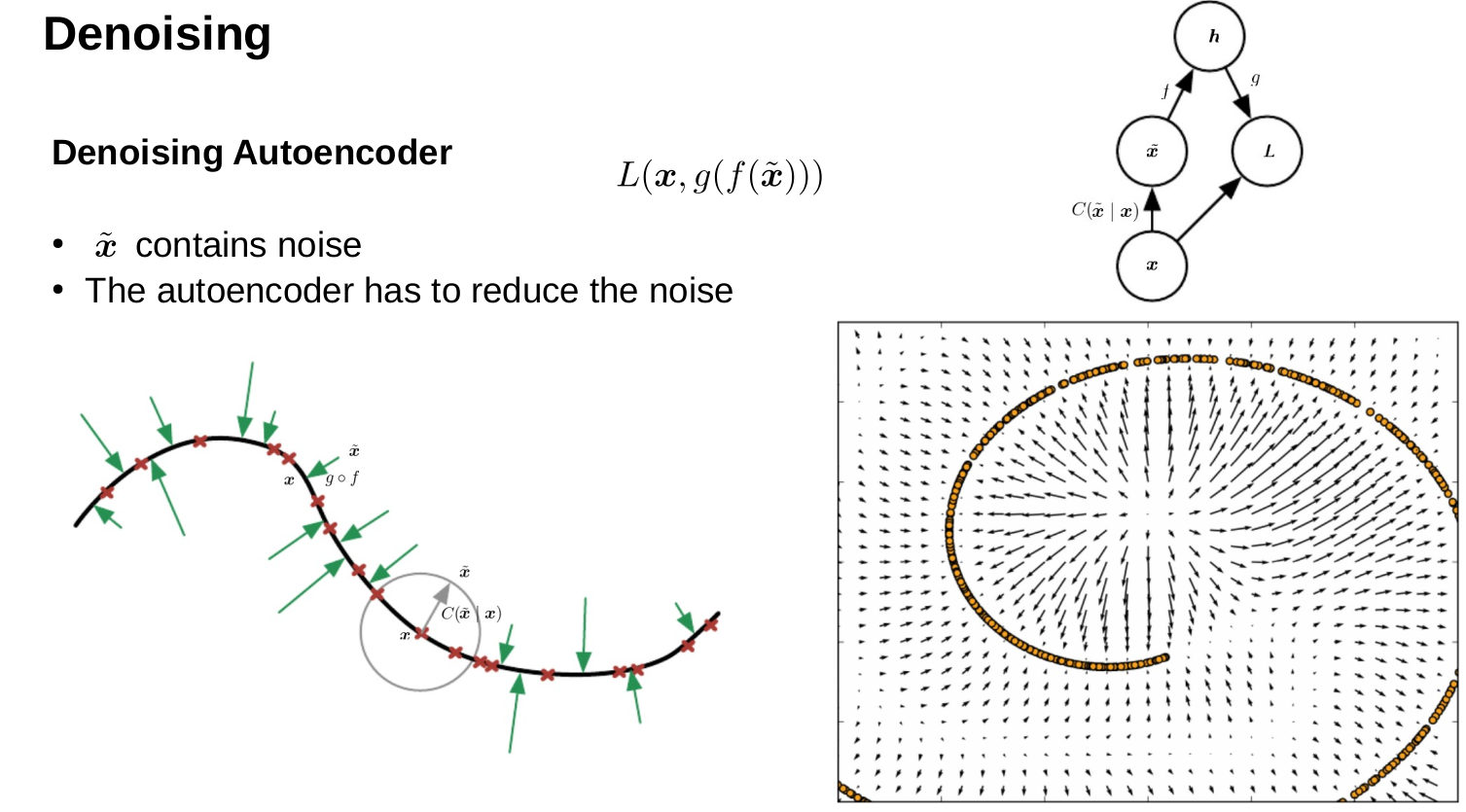
Denoising Autoencoder
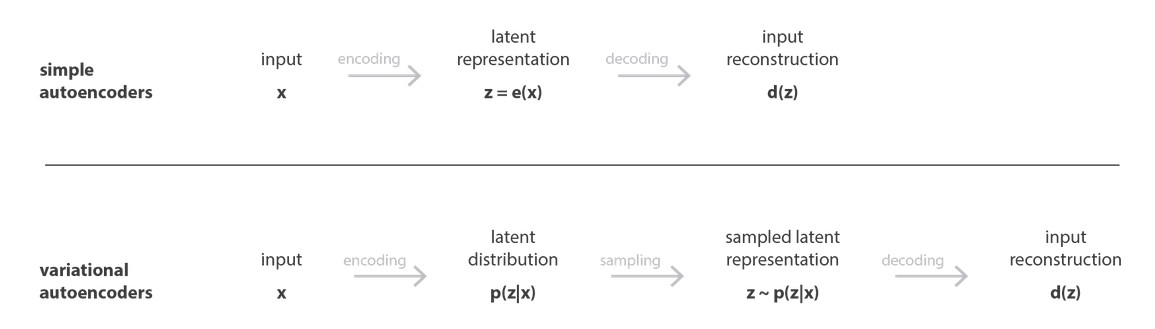
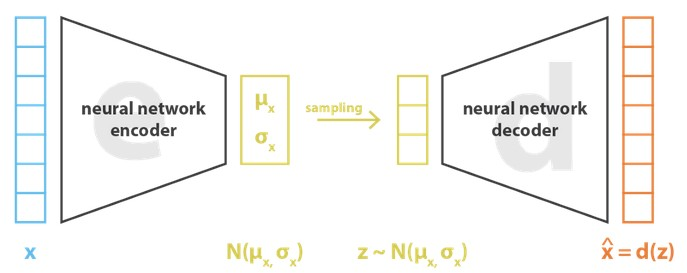
Variational autoencoder idea
Idea: Use a probabilistic model to describe the latent space
How to use VAE to get a molecule with desired properties.
Find an optimal point in latent space and sample molecules around that point
Bayesian methods for autonomous experiments
What are the problems of grid search for automatic experiments
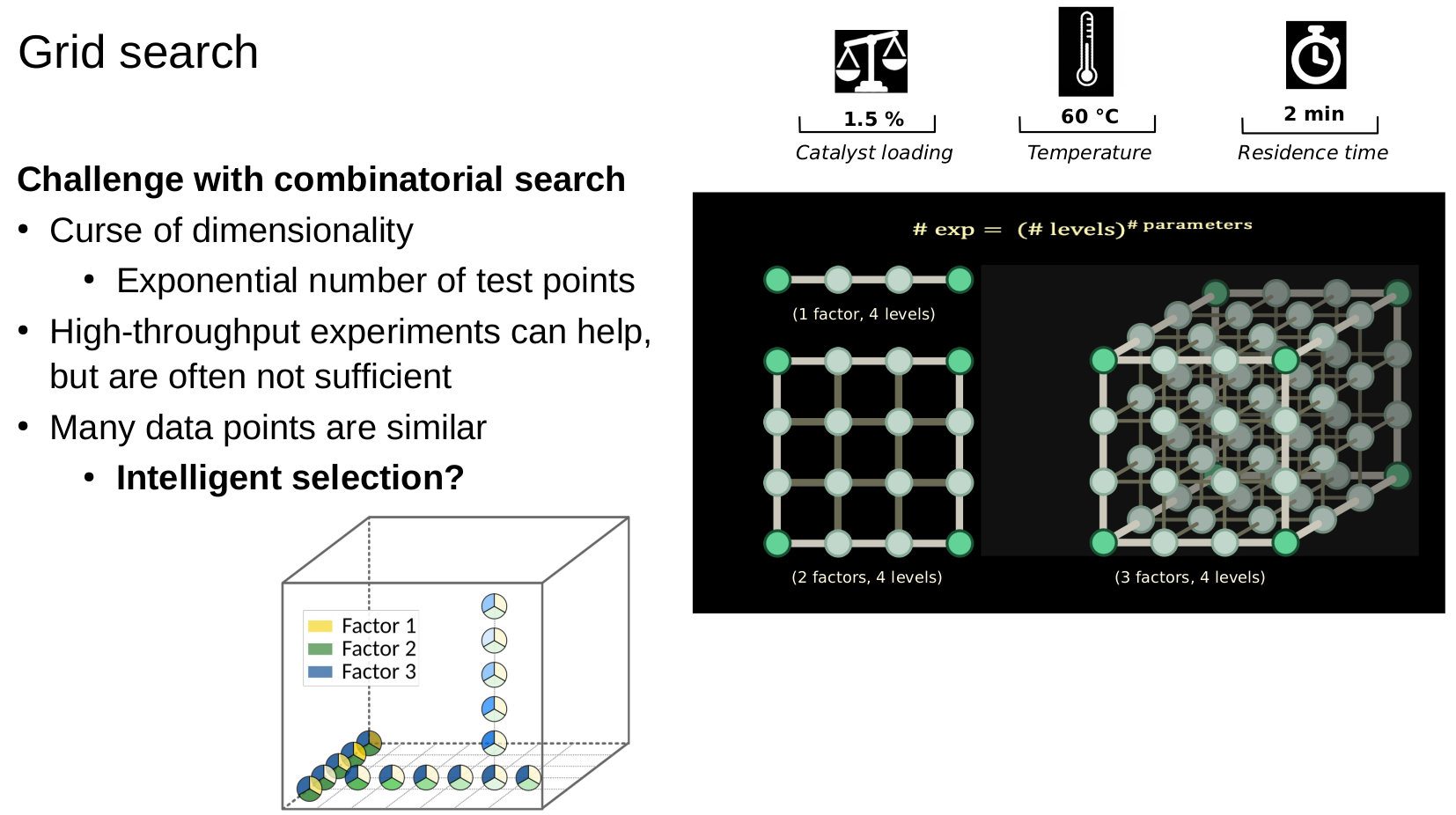
Explain the exploration exploitation tradeoff using the general form of the exploration function

Definition of expected improvement
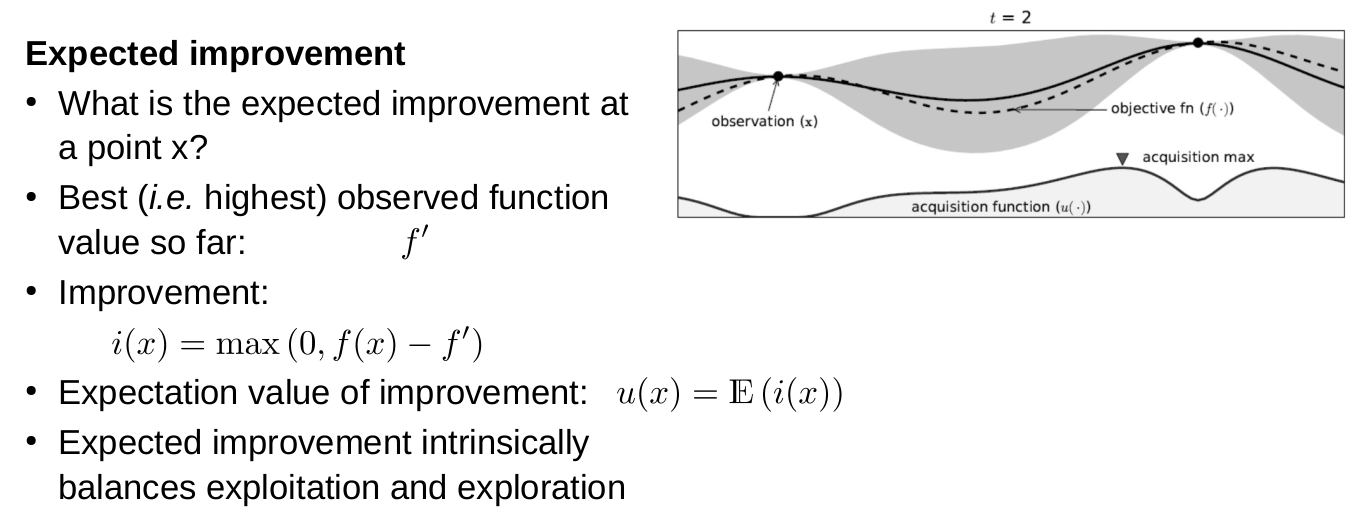
Reinforcement learning
Properties of reinforcement learning
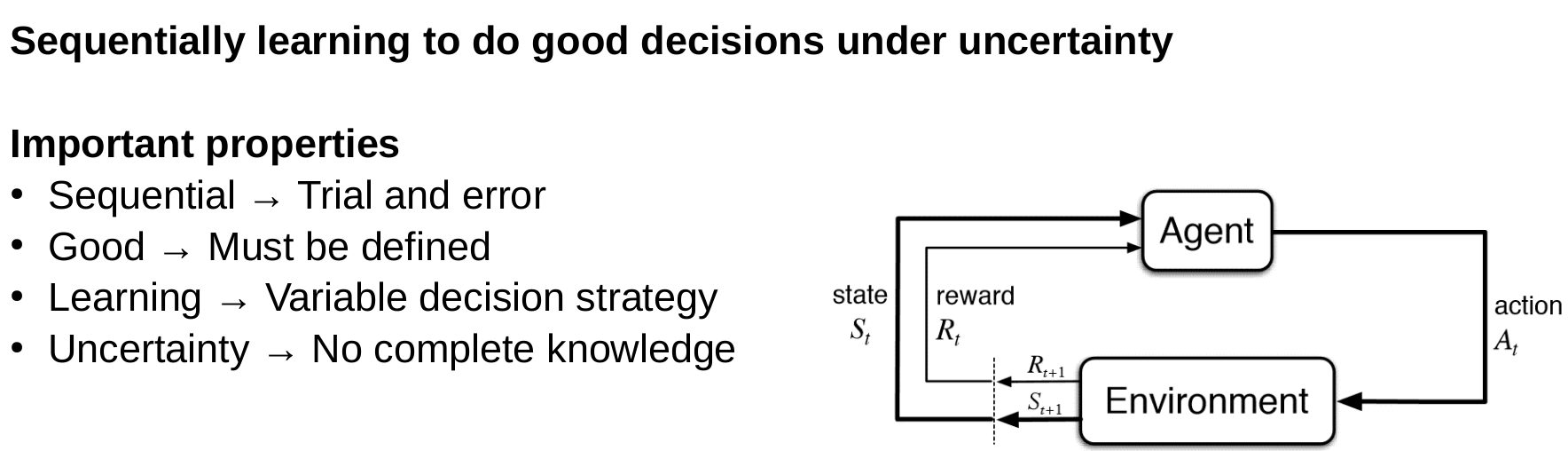
Goal of reinforcement learning
Goal: Maximization of the total expected future reward
Expected is important because the environment can be stochastic
Bellman optimality equation

Q-value update
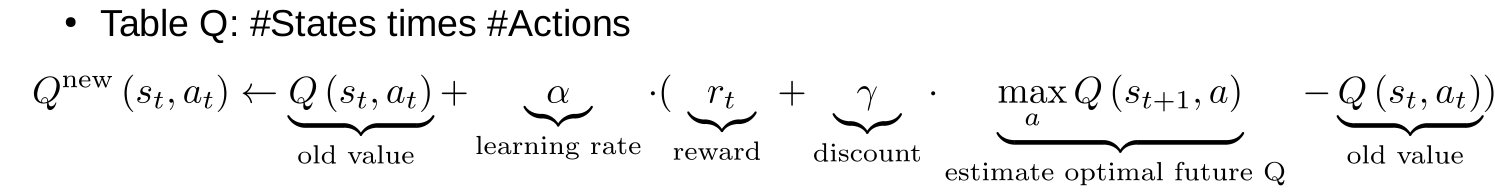
Review & outlook
- Time to read the questions
- 13:00 - 15:00
- Questions in German and English
- Take the German one and answer in a combination of German and English
- What methods for what task
- Is it sequential
- does it give us uncertainty
- What are inputs and outputs?
- How much data is needed? ⇒ learning curve
- History of AI
- Do not overhype things
- Be reasonable when communicating results: What is it useful for, what it can’t do
- What methods can be used for finding new molecules
- Which methods are supervised, which are unsupervised
- Lecture 7 discussed one application of standard FF networks for atomic movements
- Predict energies and get forces
- Lecture 9 how to replace this with GNNs
- What can be done with GNNs, but not FFNNs
- Properties prediction
- What can be done with GNNs, but not FFNNs
- Lecture 11 and 13 are not concerned with predicting the energies and properties of existing molecules, but for constructing new molecules
- What is a ROC curve!
- There is one question about the gated recurrent unit (that was more present in the exercise)
- You don’t need 100% to get the 1.0. Broad preparation pays off
- No matrix derivatives
- no backpropagation
- 80 points overall, largest question with a,b,c,d,e is 5 points so a lot of questions, pay attention to the operators
- go through the slides look at the main takeaway messages
- like why are CNNs and not FFNNs used for images
- no questions that are only about the domain
- no questions about the specific datasets
- grade average of last year will be put on ilias
- the exam is curved!
- no derivation of linear regression, but maybe intuition like we want to minimize MSE by taking the derivative and know that there is only one minimum, so we don’t need to take the second derivative
Example Questions
Level 1 (knowledge), topic "decision trees and ensembles", type "free text, methods": Q: List the steps of the greedy training algorithm for decision trees.
Recursive and greedy algorithm:
- Input: Labeled data
- For all attributes and all attribute values: Find the best split (by sorting and splitting) according to some metric like gini index, split entropy or minimum classifiction error
- Repeat this for the two splits recursively with the not yet used attributes
- Until all nodes have the same label or a prepruning stopping criterion is reached or there are no more attributes to split on
Start from the root node, i.e. the entire training data set. Evaluate the impurity reduction of a given node for all features and all possible splits. Pick the feature and split with the highest impurity reduction, split the dataset accordingly, and proceed with the two resulting child nodes according to the same algorithm. Stop when the child nodes are pure or when a convergence criterion is reached.
- Level 2 (comprehension), topic "CNNs", type "free text, methods": Q: What are the components of a ResNet block (drawing is encouraged)? What is the purpose of skip connections in the ResNet architecture?
- Level 3 (application), topic "decision trees and ensembles", type "derivation" Q: Assume you are training a decision tree using a greedy training strategy and n data points each with k features. Derive and explain the average scaling of the runtime of the algorithm in n and k to train the root node? What is the average depth of the full tree? How does the runtime scale to train the full decision tree (ignore constant prefactors)? Explain all answers.
Level 4 (analysis), topic "GNNs and molecular representations", type "free text, applications": Q: You have a very large dataset of molecules and their properties, e.g. their toxicity. You want to train a machine learning model to predict that property. Rank the following ML models according to their expected accuracy, and explain your ranking: a) Feed-forward neural network with molecular fingerprints as input vectors b) Graph neural networks which learn directly from the adjacency matrix and the molecular geometry c) Easy-to-compute molecular features concatenated to a feature vector used as input for a random forest regression model